

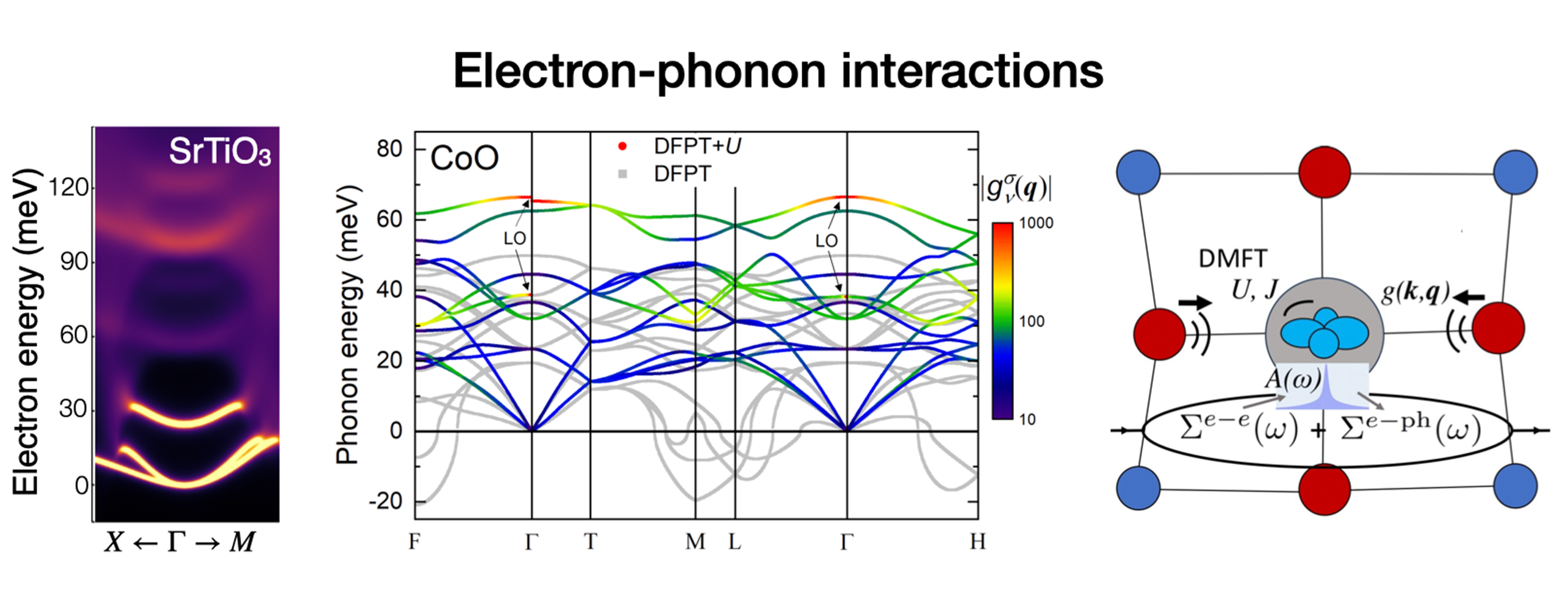
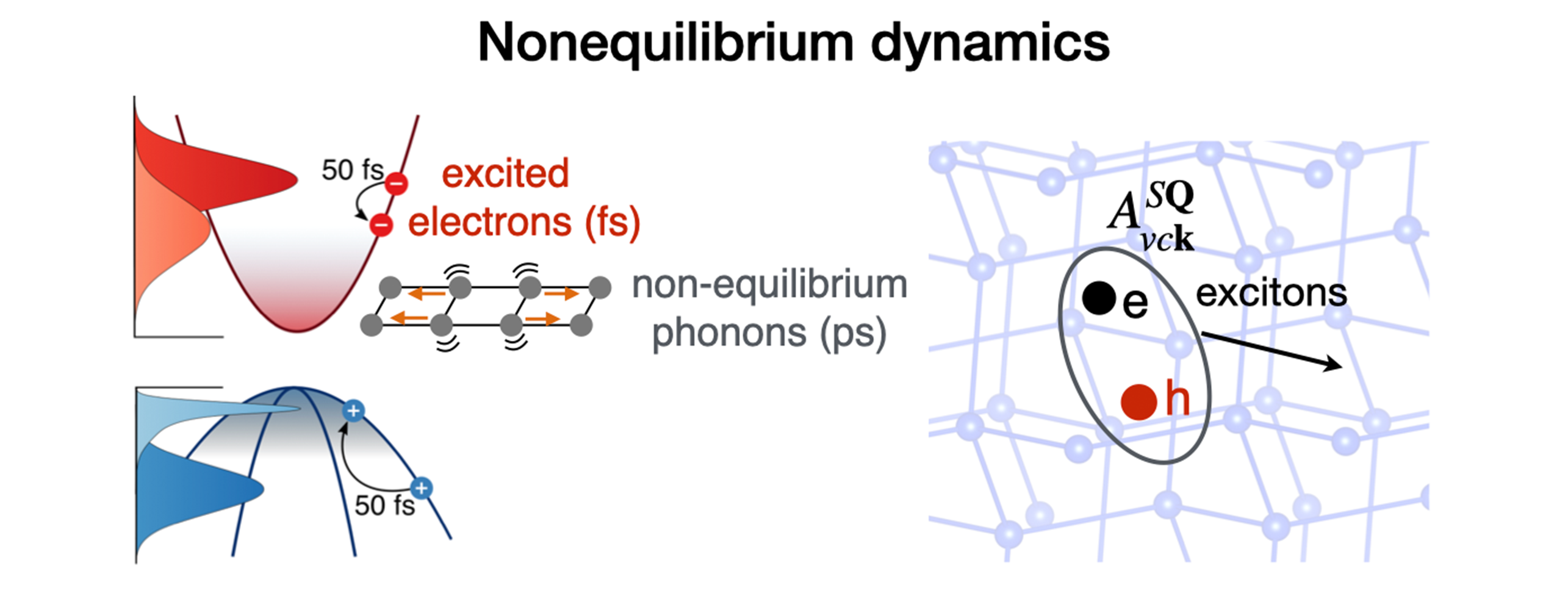

Our group develops theory and computational methods to study the behavior of electrons in materials.
We focus on first-principles calculations, which can predict the properties of materials using numerical quantum mechanics without any input from experiments. Our research sheds light on the quantum interactions and dynamics of electrons, atomic vibrations, spin, and other excitations in condensed matter.
Knowledge of these microscopic processes advances the understanding of transport, nonequilibrium dynamics, spin physics, and light-matter interactions. This work spans a wide range of conventional and quantum materials with both fundamental interest and technological applications.
We have openings for graduate students. Please contact Prof. Bernardi to discuss.
Recent News
- We show a machine learning technique to compress phonon interactions and dramatically speed up their calculation. See the paper in Physical Review Letters and the story from Caltech News: New AI Technique Unravels Quantum Atomic Vibrations in Materials. 9-16-25
- We solve the polaron problem in real materials by developing first-principles diagrammatic Monte Carlo calculations. Read the article in Nature Physics and stories from Caltech news and Physics World. 7-15-25
- Marco gives an invited talk at the KITP on our recent work on strong coupling and compressing interactions in matter. The talk is available online on YouTube. 2-3-25
- We report a technique to compress electron-phonon interactions and greatly accelerate their calculation. Read the paper in Physical Review X and the story from Caltech News. 6-1-24
- Our paper on electron-phonon interactions in strongly correlated materials is featured as Editor's Suggestion in Physical Review Materials. 9-9-23
Recent Publications
- Magnon-phonon interactions from first principles.
Physical Review B 2025 112, L180403.
- Tensor learning and compression of N-phonon interactions
Physical Review Letters 2025 135, 126101.
- First-principles diagrammatic Monte Carlo
for electron-phonon interactions and polaron
Nature Physics 2025 21, 1275 - Advancing simulations of coupled electron and phonon nonequilibrium dynamics
using adaptive and multirate time integration
npj Computational Materials 2025 11, 256
- First-principles electron-phonon interactions and polarons
in the parent cuprate La2CuO4.
Physical Review Research 2025 7, L012073 - Respective roles of electron-phonon and electron-electron interactions
in the transport and quasiparticle properties of SrVO3
Physical Review Letters 2024 133, 186501. -
Data-driven compression of electron-phonon interactions.
Physical Review X 2024, 14, 021023.